Educational content on VJHemOnc is intended for healthcare professionals only. By visiting this website and accessing this information you confirm that you are a healthcare professional.
 A three-part session featuring MDS experts Amer Zeidan, Amy DeZern, Michael Savona, Mikkael Sekeres and David Steensma, who discuss the latest updates in myelodysplastic syndromes (MDS).
A three-part session featuring MDS experts Amer Zeidan, Amy DeZern, Michael Savona, Mikkael Sekeres and David Steensma, who discuss the latest updates in myelodysplastic syndromes (MDS).
![]()
A three-part session featuring MDS experts Amer Zeidan, Amy DeZern, Michael Savona, Mikkael Sekeres and David Steensma, who discuss the latest updates in myelodysplastic syndromes (MDS), including clinical trial participation and endpoints, drug approval as well as the latest data and future outlooks.
Clinical Trial Participation
“We actually did an analysis, grouping data from five of the centers that care for a lot of patients with MDS, and only a little bit over 20% went on an interventional trial. The patients tended to be living a little closer to the center, they tended to be from wealthier zip codes, surrogate for incomes. So, there are certainly biases in who goes in study.”
– David Steensma
Endpoints & Drug Approval
“I think the way we structure clinical trials, to exclude a lot of the patients who are our target population, has really hurt us over time. And there are two lines of argument with this; one is that we should wait until we have a great study, with a clinically meaningful endpoint, like survival. Or some improvement of patient-reported outcomes, which is incredibly hard to show in clinical trials…
Another approach, one that Ken Anderson has actually promoted in myeloma, is you keep approving these drugs that aren’t that excited, with little, tiny, incremental benefits. And one day you’ll be like multiple myeloma, where all patients are all of a sudden living for years because of those incremental benefits.”
– Mikkael Sekeres
Latest Data & Future Outlooks
“I worry a little bit about the pace at which these oral agents are being approved. They’re readily accessible for pretty much everyone. And just because something is a pill does not mean it is not toxic. Some of these have very significant potential side effects, that if you’ve seen them you can manage proactively, and hopefully mitigate some of that toxicity. But I think there is a sincere learning curve”
– Amy DeZern
Amer Zeidan:
Hello. My name is Amer Zeidan. I’m an Associate Professor of Medicine at Yale University, and the Director of Hematology Early Therapeutic Research. And it’s a pleasure here to be with you in the first episode of the series called MDS talks, with VJHemOnc. In which we are talking about MDS, the latest developments as well as many issues that pertain to MDS. From clinical trial design, from latest developments in the understanding of the disease, and the genetic changes. As well as patient perspective, and different studies that apply to the disease.
Amer Zeidan:
It’s a pleasure to have such a great group with me, in this first iteration of this series. So, today I have with me Amy DeZern, who is an Associate Professor of Oncology and Medicine at Johns Hopkins University. I have Dr. Michael Savona, who is a Professor of Medicine and Cancer Biology, and the acting Chief of Hematology at Vanderbilt University. I have Dr. Mikkael Sekeres, who is a Professor of Medicine at Cleveland Clinic, and Director of Leukemia section over there. And soon to be the Chief of Division of Hematology, as a Sylvester Cancer Center in the University of Miami, congratulations, Mikkael.
Mikkael Sekeres:
Thank you.
Amer Zeidan:
And lastly, we have… last and not least, for sure, we have David Steensma, who’s an Associate Professor and the Edward Evans Chair of Medicine at the Dana-Farber Cancer Institute. Thank you so much all for being with me today, and it’s a pleasure to have all of you.
Amer Zeidan:
So, maybe I can start with David. David, in MDS, I think, historically we had a lot of issues with clinical trial participation. And this has, I think, somewhat impeded the development and introduction of new therapies. You and several, I think, investigators have looked at issues related to clinical trial participation, specifically within the MDS world. So, I was hoping you can kick this off for us, by talking about some of those main points that you have concluded from your work.
David Steensma:
Yes, I think that’s a good place to start, Amer, because we’re not going to get any better at treatments for MDS, unless we do clinical studies in patients enrolled in them. And it is hard to do MDS clinical studies; many of the patients are older, some of them have had other cancers, and may have complications from treatment of those, or may even have active disease. Older patients often don’t travel very well, or have comorbid conditions that make them ineligible for trials. And just in general, there aren’t enough studies out there for patients with MDS, particularly in smaller centers in the community, where most patients are seen.
David Steensma:
We actually did an analysis, grouping data from five of the centers that care for a lot of patients with MDS, and only a little bit over 20% went on an interventional trial. Even of those major centers. And the patients that did go on trial, they tended to be living a little closer to the center, they tended to be from wealthier zip codes, surrogate for incomes. So, there are certainly biases in who goes in study.
David Steensma:
And one last thing I’ll say, is often the eligibility criteria in the studies are way too strict. Much stricter than they need to be. Mikkael has written about this, and actually he and his colleagues at Cleveland Clinic have some data about things, like minor LFT abnormalities, or exclusions for therapy related disease that are really not necessary, and I think are a barrier. So, it is an important problem that we need to address as a community.
Amer Zeidan:
So, maybe taking it from this point, Mikkael, can we expand more on what do you think are some of the ways to try to modify these trials, in a way that would allow more participation? I think it’s quite… One of the first things that I was surprised when I saw that report in Cancer, was even in big centers which are deeply invested in clinical research, that the participation rate remains low.
Mikkael Sekeres:
Yeah, I think there are a couple of issues with it Amer, as David so eloquently already said. The first is that the eligibility criteria are overly stringent to enroll particularly typical patients we see with myelodysplastic syndromes. These folks are older, the median age at diagnosis is 70 or 71 years. In states like Ohio, they have a lot of comorbidities. So, it’s the rare patient I see who doesn’t have heart disease, or some degree of obesity, or diabetes. And we’re constructing trials with an endpoint of getting a drug approved. Which is an end point that’s defined by the sponsors of those trials. But that’s not the endpoint we care about. The endpoint we care about is getting our patients better.
Mikkael Sekeres:
And I think we’ve been complacent in allowing trials to be designed for a perfect population, through a regulatory lens. As opposed to the population we have to sit three feet away from every day, and talk about whether or not a treatment is going to work for them.
Mikkael Sekeres:
So, some of the areas in which the eligibility criteria are too rigorous; we actually did a study looking at 100 randomized trials in hematologic malignancies, published over a five-year period. And found that the eligibility criteria had little to nothing to do with known adverse events from drugs, from earlier phase trials. Nor realized events on those trials, with the exception of exclusion criteria for neurologic toxicities. So, these included liver, kidney and cardiac abnormalities.
Mikkael Sekeres:
We then took this to the next step, where we looked at studies that were conducted through the NCI Southwest Oncology Group over a 10 year period, in leukemia. There were a total of about 2,500 patients who enrolled on to those trials. And a posteriority, so after the fact it was found that some of those patients had been ineligible for those trials. But people didn’t know it, because the trials weren’t audited until later on. Well, we looked at those ineligible patients, who once again comprised about 10% of patients enrolled on those trials, and found that they had the same rates of serious adverse events as patients who were eligible, and the same remission rates as patients who were eligible. In other words, it was proof principle that the eligibility criteria were overly stringent.
Mikkael Sekeres:
Now, a lot of those patients would have been found to be ineligible really for timing issues. So, for example, maybe a study required they have a bone marrow biopsy within two weeks of enrollment, and they had a bone marrow biopsy three weeks from enrollment, therefore should have been ineligible. And these are nonsense criteria, that we can modify to be overly inclusive of patients in those trials. So that the trials better reflect the patients we’re going to have to face one day. And think about offering a drug, once it’s approved.
Amer Zeidan:
Yeah, those are great points. Amy, maybe I’ll take it to another dimension. Both us trained at Hopkins, and I think one of the things that struck me during my time there, when we were talking to patients about clinical trials, is some degree of some maybe distrust of the system. That comes to some of the history of how clinical trials, or clinical research in general has been done in countries, including the US. And maybe what some of the minority populations, these issues have been more, I think, of concern. And the issue that patients bring up often, about not wanting to be a guinea pig.
Amer Zeidan:
So, what do you typically do, when you have those difficulties in talking to the patient? You make the trial appear more reasonable to them, about something that they should really consider?
Amy DeZern:
So, it is a complicated question. And you’re right, both in Baltimore and in other parts of the US, and all over the world. It is an opportunity, as opposed to a difficulty, to really ensure that we provide truly informed consent. If we’re trying to convey to a patient that it’s quite important that we move them beyond the current standardly available therapies, to try and give them the best opportunity to have a better outcome with their disease. And I find that this is something that I welcome the opportunity to do. It takes a lot of time. And the term guinea pig does come up in my clinic visits more than I would have ever anticipated when I was in medical school.
Amy DeZern:
And I really think that the onus is on the provider, as a clinical investigator, even the principal investigator of the trial, or just a co-investigator. To really make sure that we understand the mechanism of a drug, why it makes sense in this individual human being, and then what the alternatives are for that particular patient’s therapy. Because there really are some patients where I’m not comfortable as their doctor to offer them a randomized trial, where they could receive placebo, because I think that there might be something else better out there. And so there are some patients where we have to just really weigh the risks and benefits of standard therapies, and how likely they are to work. And then for the investigational product, how toxic it might be, and if that fits with the patient’s roles for the treatment of their particular disease.
Amer Zeidan:
Yeah, those are great points. Maybe, Michael, I think another dimension for the clinical trial participation is, especially in states where they are not as densely populated, where many of the patients live far from the big center, and they have to drive one or two hours basically, to come to you to do a clinical trial. What are some of the, I think, some of the challenges that you’ve foreseen, and how do you try to work with patients to try to make some trial options available for them that they can participate, even if they live far from here?
Michael Savona:
Well, Amer, I mean, I’m an optimist. So, one, the flip side, the silver lining of there being such a lack of good therapies for MDS is that there’s a lot of opportunity. There’s a lot of opportunity in clinical trials to test new mechanisms of action, which we’re doing, and have made progress in the last several years. But also finding ways to deliver the things that we know work, to patients in a more efficient and effective manner.
Michael Savona:
Here in the South, and in Tennessee, we have patients who are not just an hour, but three, four, up to six, eight hours to come into a major medical center. And sometimes their closest oncology office is as much as an hour and a half, to two hours away. In those cases, coming to sit in an infusion chair and receiving an IV chemotherapy for consecutive days on end, for example, is not appealing.
Michael Savona:
So recently, we were involved in developing a oral analog of decitabine, which is a common DNA methyltransferase inhibitor, used in the standard care with MDS, and there’s an oral analogue given with the drug, called cedazuridine. Cedazuridine blocks cytidine deaminase, which is a key enzyme in the gut and the liver, that breaks down decitabine. So, oral decitabine alone, by the time it gets to the blood stream, is not a bioequivalent drug. But given with cedazuridine, the bioequivalents could be matched. And through a series of pharmacokinetic-driven experiments, we were able to show that basically cedazuridine and decitabine, in a fixed dose combination of 35 mg of decitabine and 100 mg of cedazuridine is really the same as IV decitabine.
Michael Savona:
So now, patients can be given a… See the doctor on day one, and instead of coming back days one through five, every day for parenteral administration of decitabine, they can get their pills and go home. So, it’s not as exciting as far as a novel mechanism of action, but it’s very important for improving quality of life. This is not a disease that we’re able to cure the vast majority of patients, with allogeneic stem cell transplant. So, improving quality of life and adding good years, not just years, for patients is really the motivation behind a lot of these types of developments.
Amer Zeidan:
Yeah. And that actually brings me to the next topic I wanted to discuss. Since you brought up the oral decitabine, and this Phase III, the ASCERTAIN trial. And I think the novel way of doing the endpoint. I think Mikkael touched a little bit on the subject of the endpoints of the trial, and this Phase III trial of the pharmacokinetic equivalence between the oral and the IV versions of decitabine.
Amer Zeidan:
So, I think we historically have this disconnection in MDS, between the short-term endpoint, such as hematologic improvements, or complete response and the long term outcomes, because they don’t always correlate. We don’t have that same degree of correlation between progression-free survival and overall survival, that are seen in some sort of tumors. And I wonder, from both the patient perspective, as well as a regulatory perspective… maybe I can go back to Mikkael and David, especially since both of them sat on the ODAC, helping the FDA to decide on some of those drugs.
Amer Zeidan:
And we are… probably things are changing these days in terms of how drugs are being more approved somewhat faster. But when you look at the current endpoints, many of the trials for MDS are using CR as an endpoint. So, what’s our perspective on this? Maybe I can start with David, and then go to Mikkael.
David Steensma:
At the end of the day, what matters most to patients is living longer and living better. So, better quality of life, patient recorded outcomes, minimal burden, such as transfusions, but then living longer. And in so far as their blood counts correlate with those things, or a decrease in blasts correlate with those things, then those are beneficial endpoints.
David Steensma:
I think things like hematologic improvement, while they can be important, for instance, if they decrease a patients transfusion needs, or help them have more energy, or bleed less, can be meaningful. But minor improvements in counts really probably aren’t that helpful for patients.
David Steensma:
What is a CR worth? Well, I think if it’s a true CR, decrease blasts and increase counts, then that does seem to correlate with longer survival. One always has to be careful about assessing survival based on response, because it can be confounded unless you do a proper landmark analysis. People still do these things, we’ve known about this since Anderson’s paper in the JCO in 1983, that that was a problematic way of doing things.
David Steensma:
Marrow CR, I think we’re not as sure that that’s a meaningful endpoint. If the blasts go down, but there’s no count recovery, is that really helpful for the patient? And for instance, rigosertib is a drug that quite commonly decreases the blasts, but now we have two negative studies with rigosertib against conventional care. And so that doesn’t seem to translate into improved survival.
David Steensma:
So, I think we are seeing more use of these surrogate endpoints. And they’re not super well validated. I will say this is happening across disease types, so it’s not specific to MDS, that we’re seeing things approved on PFS endpoints, or other surrogate markers.
David Steensma:
When we were in ODAC together, I was like the good cop, and Mikkael was like the bad cop who would do the real interrogation, and I’d try to give the guys a way out, but Mikkael would go for the jugular. So, I’m curious to see what he would say.
Mikkael Sekeres:
Well, I want to point out, that with Anderson’s article in 1983, David actually read that at age 12, when it first came out. While I was wasting my time being terrified of talking to girls.
Mikkael Sekeres:
So, I still struggle with where we’re going with regulatory endpoints, and how we’re designing trials in myelodysplastic syndrome. And I don’t think I have the right answer. I joke with people, I wish I had a home chemistry kit, because the rate at which FDA is approving drugs based on interim markers of clinical benefit, or markers that may not even be interim markers of clinical benefit, I could get something approved pretty quickly.
Mikkael Sekeres:
The drugs that we had approved for MDS this year are okay. I think, as Michael alluded to, and he was instrumental in getting one of those drugs approved, they’re not exciting nor game changers. There is something else to do. And we’re still waiting for that game changer. One of the challenges with doing it, is that I think we’re lackadaisical about enrolling patients onto clinical trials. And I think the way we structure clinical trials, to exclude a lot of the patients who are our target population, has really hurt us over time. And there are two lines of argument with this Amer; one is that we should wait until we have a great study, with a clinically meaningful endpoint, like survival. Or some improvement of patient-reported outcomes, which is incredibly hard to show in clinical trials. And we shouldn’t budge until we get that trial. Another approach, one that Ken Anderson has actually promoted in myeloma, is you keep approving these drugs that aren’t that excited, with little, tiny, incremental benefits. And one day you’ll be like multiple myeloma, where all patients are all of a sudden living for years because of those incremental benefits.
Mikkael Sekeres:
As David alluded to, I’m in the first camp, where I think we should wait for studies with meaningful endpoints. Because until then, we’re hawking false hope to our patients, instead of true hope and true progress in their disease. But I readily admit that there are other ways of thinking about drug approval.
Amer Zeidan:
So, Michael and Amy, I’ll go to you with this question. So, David and Mikkael actually wrote one of my favorite editorials that I quote often, they call it “the boulevard of broken dreams” about drug development for AML. And how many drugs were basically being disapproved, and I suspect, and I think they would probably agree with me, that if that decitabine trial that was not approved back in 2012, if this was brought in front of the FDA now, that might be a different consideration.
Amer Zeidan:
And I wonder, are we going to a direction where we are really having some major breakthrough in our drugs for MDS, or is it that the approval considerations have changed? So, in terms of what Mikkael was talking about, approving a number of drugs that have small, incremental benefits, but overall can’t change a natural history of disease. Versus trying to go after drugs that are major game changers, and how we manage those patients. What’s your thoughts on this, maybe start with Michael and go to Amy?
Michael Savona:
Well, I was going to say ladies first, but I’ll chomp at the bit, since you asked me. I think Mikkael is right, there’s a sacrifice to be made. If the approach of approving a lot of therapies, to actually get them out there, there are a lot of patients who are not going to benefit by those things. But in the end, 80% of childhood acute lymphoblastic leukemia is cured with chemotherapy, because of that trial and error step-wise approach that was used.
Michael Savona:
Now, the other issue, just to play devil’s advocate, is that MDS is myelodysplastic syndromes, it is many, many, many diseases, as is AML. It’s a very heterogeneous disease. And we have plenty examples of quote unquote “loser” trials, that did very well for some patients. But those patients were not like everyone else in the trial. So, one would argue that the paradox that we have to deal with, is that the diseases we’re studying are more and more and more different from… and sub-groups within them, yet we have to have numbers great enough to actually have a statistical, meaningful endpoint. So, you end up having some kind of homogenization there.
Michael Savona:
I think that there is something to be said for the approach that has been followed in myeloma. These incremental benefits now… When we all started practice, people lived for a median survival, about three to five years with myeloma, and now people are living a decade or more. And that’s because of the developmentic and new mechanisms of action, but also further exploration with a proven mechanisms action. To now, where we have 15, 20 drugs to use, that just didn’t exist 20 years ago.
Michael Savona:
So, I think that in MDS, none of the drugs that are approved this year are game changers on a population scale, but are meaningful to some patients. And finding ways to identify which patients will respond to therapies quicker is important. And that’s actually the model in my laboratory, finding the best therapies for the right patients, as quickly as possible. Because really, what we’re doing in science is all about moving this quickly to patients, and providing value to the folks we take care of in the clinic.
Amy DeZern:
Sure. Just to let the ladies have the last word. I’ll mention that… drawing together a lot of the themes that the previous three gentlemen have mentioned. I don’t necessarily think we have to pick between the be all, end all, overall survival trial and then trials that have more modest endpoints, that do, as people have alluded to, have individual, incremental benefit for an individual patient. I actually think the heterogeneity in the disease overall, that Dr. Savona alluded to, was probably one of the most critical things to understand. We still are fairly nascent in our understanding of the biology of all of these syndromes overall. We’ve made tremendous headway over the past 10 to 15 years, but I think we still have some room to go.
Amy DeZern:
And I don’t think… When I plan my personal MDS portfolio here at Hopkins, I like to have a blend of both of those type of trials, where we can. Quite often, the overall survival trials are the ones I look for for the higher risk, higher reward. But for the more indolent, lower risk disease, I think that incremental benefit for a chronic condition for these patients, who are more advanced in age, really also has benefit. And so I try and keep it balanced, in that way. And think about regulatory approvals that fit the whole umbrella of the myelodysplastic syndromes.
Amer Zeidan:
Yeah. And I think, when we think about all of these new drugs… And probably not for the sake of time today, I might not go into all the details of the new agents. But I think from a mechanistic point of view, historically with MDS, many of the drugs that we have approved, we generally do not fully understand the mechanism. We probably still continue not to fully understand the mechanism, whether it’s the hypomethylating agents, whether… lenalidomide took several years to fully understand. And each, two, three years we get a new potential mechanism of action.
Amer Zeidan:
And I wonder, now that we have so many different agents, how do we go about trying to get all of these different drugs that are being currently tested… we have a large number of Phase II and Phase III trials that are going forward. And each sponsor is doing their own large Phase III trial. And there are not many patients to go on to all trials. And I think Mike mentioned that there are a lot of probably differences between the patients, I mean, not every high risk MDS patient is the same as other patients.
Amer Zeidan:
So, how do we go about trying to do a more rational approach of designing and integrating these trials? Maybe I can start with David, citing the experience of the spliceosome inhibitor? I think there was a lot of excitement, in terms of this pathway. And again, we thought that we fully understood how this works, and many of us were somewhat… I’m sure you were one of them, David, were disappointed with the clinical results. With… at least initial clinical results with this agent. What do you think of the connection between the biology and the selection of the drugs, and the design of the trials should be, to have the best outcomes for patients?
David Steensma:
This is a difficult issue, that we’re wrestling with. And I think our acute leukemia, and many of us also do see patients with acute leukemias, and the acute myeloid leukemia experience has illustrated this. So, there are some agents, like FLT3 inhibitors, or IDH1 and 2 inhibitors, that are targeted, where maybe it’s a little clearer who’s the optimal patient for that. But then there’s other agents, like glasdegib, where it isn’t as clear. And now that we have a positive overall survival trial with azacitidine plus venetoclax, now do we need to recombine each of those drugs now with aza-ven or decitabine-venetoclax, that were previously started with azacitidine alone in combination. Aza-ven is not yet a standard of care in MDS, but it certainly could be, and is going to make that more complicated.
David Steensma:
The meaning of the drugs that are moving forward in MDS, we don’t have a specific patient subgroup that is going to be a responder. So, pevonedistat, the TIM-3 inhibitor, CD47 inhibitors like magrolimab. There’s a few exceptions, like the APR-246 drug, which is TP53 directed, but other agents also are active in TP53 mutant patients. So, these are, I think, some of the challenges that we are wrestling with as a field.
David Steensma:
And you’re right, Amer, it’s often there’s a great story behind why a drug should work. And then it turns out not to be as efficacious as we thought. And H3B-8800 is a good example. Omar Abdel-Wahab had a very encouraging preclinical data, the study we did didn’t show any CRs. There were some patient who became transfusion free. Why is that? Were we studying the wrong… Is it the molecule, is it the dosing schedule? And there’s going to be some follow up on that, with some new dosing schedule.
David Steensma:
So, this has happened again and again in our field. We still don’t know exactly how hypomethylating agents work. And why there are these patients who are super responders. I have four or five patients in my practice that are out more than five years and still in CR, on these drugs. But what’s special about them? Compared to all the others who didn’t respond, or responded for a year, and then lost response. We don’t have good markers at the moment to help us know who those are going to be.
David Steensma:
But we think we know lenalidomide works, Ben Ebert’s work really sorted that out nicely, in the del(5q) subset. But we don’t know how it works in the non del(5q) patients, when it does. So, yeah, I think it’s messy.
Amer Zeidan:
Yeah. And just to follow up on the same point, Mikkael, you led one of the larger trials basically around the mice trial of aza, versus aza with vorinostat, versus aza with lenalidomide. I think it was, this work, I believe 1107 trial, basically. And I think one of the concerns, in addition to the points that David mentioned about the disconnection sometimes between the biology and the efficacy, is how good are the physicians about managing patients? And I think one of the issues that came up during the analysis and the discussion of these trials, where there sometimes are drugs being discontinued so quickly, are we not very good at managing side effects? And how do we do the trials in a way that does not lead to a potentially negative signal when a drug has potential activity?
Amer Zeidan:
What did you learn in the context of conduction of this trial? And how this experience can be moved to help with all these trials that are currently happening?
Mikkael Sekeres:
It’s a great question, Amer. There were a lot of things, I think, we learned from that trial. When we analyzed the data, we found that patients who received combination therapies compared to aza monotherapy therapy were significantly more likely to discontinue therapy because of toxicities. Despite the fact that the rates of grade three or higher adverse events actually weren’t higher in the combination arms than in the monotherapy. And that they were significantly more likely to undergo non-protocol defined dose modifications.
Mikkael Sekeres:
And in real time, this meant that I would get an email from somewhere in America, where someone was being treated. With a message, “You know, my patient had the side effects, so we reduced the dose of the azacitidine, and then had another side effect. Should I reduce it again?” And I would say, “Wait, wait, wait. You shouldn’t have reduced it to start with, that’s not what the protocol says.” But by that point, the damage had been done.
Mikkael Sekeres:
Put this into context with some work that David and I did through the Aplastic and MDS Foundation, where we looked at reasons why patients stopped therapy. It was a fascinating study, because we asked patients and we asked a bunch of healthcare providers. And when we posed the question, “Therapy was discontinued because side effects were too severe?,” Doctors were three times more likely than patients to say, “Yes, that’s the reason.” And when we drilled down to it, therapy was discontinued because quality of life was suffering from side effects, again, three times more likely doctors would say yes, than patients.
Mikkael Sekeres:
So, does that mean that patients are looking back towards a therapy with rose colored glasses? Or does it mean that their doctors were interpreting toxicities in a way that they weren’t feeling? Or, that they didn’t think were severe enough to stop a therapy?
Mikkael Sekeres:
So, put all of this together in a basket now. We have the North American Intergroup, S1117 study, where combination therapies were discontinued prematurely, or dose reduced not by protocol guidelines. And we have a study that says that we as physicians are more likely to blame stopping therapy on toxicities than our patients are. And what that tells me is that we have an opportunity for education, with docs. And probably have to build into any clinical trial, the requirement that before any dose reduction or discontinuation occurs, that a doctor has to check with a medical monitor. And when we did this for the Asian pevonedistat study, guess what, patients, they actually stayed on therapies longer. And as a result there is actually a signal in the higher risk MDS patients that a combination seems to work better than aza monotherapy.
Amer Zeidan:
Thank you. Amy, we have now several oral agents, we have the IDH inhibitors, we have potentially venotclax. We have those oral hypomethylating agents, both of them got approved in the same year, after many years of work, one for AML and one for MDS. Do you view that we are going… probably apply for both AML and MDS. Are you foreseeing our work, where we are treating patients with high risk MDS, and all the patients with AML, with just oral therapies, where the patients are just taking pills, and coming to the office every few weeks? Is that something that going to be a reality in the next, I don’t know, 10 years?
Amy DeZern:
So, I certainly don’t have a crystal ball. But I think oral therapies for chronic diseases has long been a holy grail, specifically in MDS. I think AML is a little bit different, because of the inherent toxicities and the higher white counts, and potential for lysis. But the reality of any therapy we choose, for almost any condition, is we’re trying to extend peoples lives with quality. And I think we all know, coming to the hospital, even seeing any of us, pleasant as it might be, is still a decreased healthcare quality of life.
Amy DeZern:
And I worry a little bit about the pace at which these oral agents are being approved. They’re readily accessible for pretty much everyone. And just because something is a pill does not mean it is not toxic. Some of these have very significant potential side effects, that if you’ve seen them you can manage proactively, and hopefully mitigate some of that toxicity. But I think there is a sincere learning curve. And maybe it is the decade that you suggest, that we need to make it a reality. But certainly, for all these conditions, we hope these patients can, outside the time of COVID, go on cruises, or something that brings them pleasure. And taking their oral chemotherapy pills is a goal for that. As long as we can do it in a truly safe fashion, with adequate monitoring.
Amer Zeidan:
So, what do you think, Michael, about all of this? Do you foresee that… You work in several of those oral agents, as well. And I know you had some nice pre-clinical data’s with. So, do you think that the direction of MDS care in the future is going to be total oral therapy combinations?
Michael Savona:
So, yeah, it’s hard to argue with Dr. DeZern. She’s usually right, and she’s right again here. I mean, obviously this comes with risk. I think there’s a precedent, though. I mean, oncologists have been giving capecitabine to patients, who get 5-FU, for many years. And you take the pill home with you, and it’s chemo, it’s dangerous to take outside of the direction of your physician. And even then, there’s sometimes complications that occur that wouldn’t occur if you were being watched more closely in a hospital. But these are trade-offs we make, and this is the responsibility of doing these things. And knowing your patients, which patients are good ones to treat in this fashion, and how to go about doing this.
Michael Savona:
So, I’m less concerned about that risk. I’m more concerned about the improper application of oral DNMTIs, based on the approvals this year. CC-486, or oral azacitidine, is a drug that’s active. It’s clearly shown a survival benefit in a study that randomized patients to placebo, or oral azacitidine, with an average of only one cycle of consolidation after getting induced with cytotoxic chemotherapy. So, the bar is pretty low. There’s clearly a survival benefit.
Michael Savona:
But that study was started before all the approvals we had in 2018, which basically changed some of the potential maintenance options you may have, first of all. But more importantly, oral azacitidine, because it has to be given over 14 to 21 days, and not over seven days, like parenteral azacitidine. Because if you tried to reach bioequivalence over seven days, it just doesn’t work, because azacitidine deanimates. So, because it’s given for 14 to 21 days, the methylome is totally changed. An active drug, but not equivalent to parental azacitidine. If the approach with cedazuridine and azacitidine, which is an ongoing trial right now, that reached a bioequivalence as cedazuridine and decitabine did just recently, and lead to that approval, that’s a different story. But CC-486, or oral azacitidine alone is a different drug, and should be used in that context.
Michael Savona:
I guess I’d like to say one more thing about some comments about the other drugs that are in the pipeline. I think David was absolutely right, we have some very interesting compounds, that we’re not really sure how to use them. And some of those drugs will be approved. But often, it is said about tyrosine kinase inhibitors that there’s more papers published as a result of the discoveries made 20 years ago with TKIs, and our patients who have CML. And I’m not sure if that’s true exactly, but that led to a scientific blossoming, where in molecular medicine we know how to do things, we just didn’t know how to do all because of those earliest discoveries that were made, and the patients that were actually treated on the first generation of tyrosine kinase inhibitors.
Michael Savona:
I think in the same way, molecular medicine is blowing open how we take care of myeloid disease. Yes, there’s targeted agents, for sure, but we’re also learning about what happens inside a cell when we treat patients. If they get venetoclax, we’re actually learning what happens inside the mitochondria to make them less or more likely to respond to venetoclax, or continue to respond to venetoclax.
Michael Savona:
Those are the types of thing, I think, that are really going to push the field forward. And help us understand how to intercalate these new drugs into the paradigm. We don’t have the luxury of designing… an org start for MDS is just one line anymore, it’s very complicated. We’re getting to look like the breast cancer diagnostic trees.
Amer Zeidan:
I think that’s well said. So, I think this is such a great discussion. I think we’re running out of time, so I just want to finish by… if any of you have any last words? Particularly about how the clinical trial space really, in MDS, should be moving over the next few years? Maybe we can start with David?
David Steensma:
I think my colleagues have really summarized things well. And this is still a very challenging group of diseases. And we talked about some of the logistics, and the challenges. But it is also very exciting, there are all sorts of new drugs being developed. I think we’re just learning too about the large number of people who had clonal hematopoeisis. And about potentially being able to find out before things become more molecularly complex. We don’t know what to do with ARCH/CHIP, whatever one wants to call it, not yet, how to reduce those qualms. But maybe that’s an early intervention strategy that may help in the future.
Amer Zeidan:
Thank you. Mikkael?
Mikkael Sekeres:
There are some analogies to the COVID pandemic and where we are right now, in an… I don’t know when people would be watching this, but we don’t have a vaccine yet. So, our best strategy against COVID is prevention. And you wonder, if our best strategy against MDS, given the fairly active compounds that we have, none of which is curative, to target CHIP or ARCH, some pre-MDS conditioning, and prevent it from ever going into MDS. Thereby extending our patients lives.
Amer Zeidan:
Thank you. Dr. DeZern?
Amy DeZern:
I echo what these gentlemen have said. I think it’s an exciting time to really change the natural history of myeloid disorders in general. If we can intervene in non toxic ways earlier, for our patients. And the more patients we’re able to put on these style of trials, where we really understand the disease biology, and we monitor toxicity closely, really will advance the field. And I’m certainly looking forward to the next decade or two, where we get to do that.
Amer Zeidan:
Thanks. Michael, you get the last word.
Michael Savona:
Oh wow, that never happens. So, I think I’ll take the opportunity for a quad agreement around the concept of clonal hematopiesis. I mean, in our professional lifetimes, this is one of those once in a decade type, once in a maybe many decade discoveries. And what’s interesting is we’ve moved because of this explosion in molecular medicine beyond the molecular epidemiology, if this mutation occurs more likely in these people, at this age. To the point we actually understand some mechanism. The people get cancer because they have this, because it causes this. And as we learn more about mechanism, and this is where a lot of the fun research is happening, then we can actually start to really be thoughtful about targeting those specific mutations.
Michael Savona:
There are going to be challenges, because the more we learn, the more the nuances between different mutations are going to become the bearers. Those of us who have CHIP clinics now are currently experiencing. But I absolutely agree with my colleagues, that this disease is too complicated to end in the majority of cases, once it’s come about. We have to find a way of keeping it from coming about in the first place.
Amer Zeidan:
Mm-hmm. Thank you so much, well said. At the end, I like to thank you all for participating in this iteration of the MDS talks. And I look forward to having you in future episodes. This is such a great group, and I think we have so much to talk about, in terms of MDS and the new developments that are happening. Thank you so much.
David Steensma:
Thank you.
Mikkael Sekeres:
Thank you, Amer.
Amy DeZern:
Thanks for having us.
Amer Zeidan:
Thanks.



Disclosures
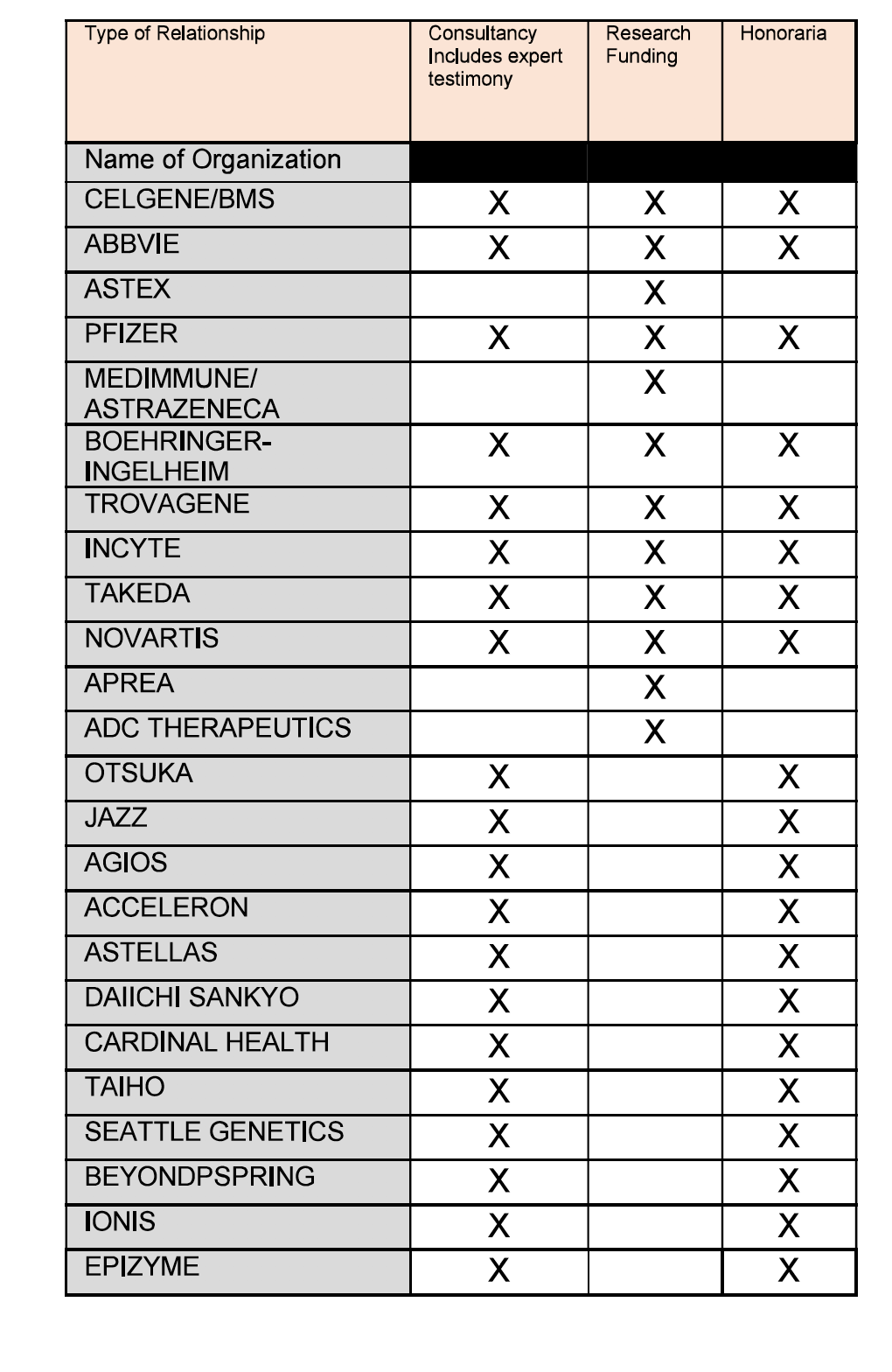{kind=link}
Mikkael Sekeres – Advisory Boards, Celgene/BMS, Takeda/Millenium, Pfizer
Michael Savona – Consultant and on company advisory boards for Karyopharm and Ryvu; has equity in Karyopharm; participated in one day advisory boards for AbbVie, BMS, and Takeda; is a consultant for BMS, Geron, Karyopharm, and Ryvu; receives research funding from Astex, Incyte, Takeda, and TG Therapeutics; and serves on the DSMBs for BMS, Sierra Oncology, an TG Therapeutics.